Documentation for Sonia package¶
SONIA is a python 3.6/2.7 software developed to infer selection pressures on features of amino acid CDR3 sequences. The inference is based on maximizing the likelihood of observing a selected data sample given a representative pre-selected sample. This method was first used in Elhanati et al (2014) to study thymic selection. For this purpose, the pre-selected sample can be generated internally using the OLGA software package, but SONIA allows it also to be supplied externally, in the same way the data sample is provided.
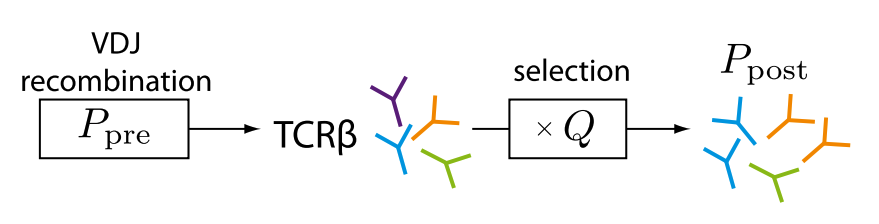
SONIA takes as input TCR CDR3 amino acid sequences, with or without per sequence lists of possible V and J genes suspected to be used in the recombination psrocess for this sequence. Its output is selection factors for each amino acid ,(relative) position , CDR3 length combinations, and also for each V and J gene choice. These selection factors can be used to calculate sequence level selection factors which indicate how more or less represented this sequence would be in the selected pool as compared to the the pre-selected pool. These in turn could be used to calculate the probability to observe any sequence after selection and sample from the selected repertoire.

Installation¶
SONIA is a python 2.7/3.6 software. It is available on PyPI and can be downloaded and installed through pip:
pip install sonia
SONIA is also available at https://github.com/statbiophys/SONIA . The command line entry points can be installed by using the setup.py script:
python setup.py install
Sometimes pip fails to install the dependencies correctly. Thus, if you get any error try first to install the dependencies separately:
pip install tensorflow
pip install matplotlib
pip install olga
pip install sonia
References¶
- Sethna Z, Isacchini G, Dupic T, Mora T, Walczak AM, Elhanati Y, Population variability in the generation and thymic selection of T-cell repertoires, (2020) bioRxiv, https://doi.org/10.1101/2020.01.08.899682
- Isacchini G, Sethna Z, Elhanati Y ,Nourmohammad A, Mora T, Walczak AM, On generative models of T-cell receptor sequences,(2019) bioRxiv, https://doi.org/10.1101/857722
- Elhanati Y, Murugan A , Callan CGJ , Mora T , Walczak AM, Quantifying selection in immune receptor repertoires, PNAS July 8, 2014 111 (27) 9875-9880, https://doi.org/10.1073/pnas.1409572111
Structure¶
Note
Note about training data preparation
Sonia shines when trained on top of independent rearrangement events, thus you should throw away the read count information. If you have a sample from an individual, you should keep the unique nucleotide rearrangements. This means that in principle there could be few aminoacid CDR3,V,J combination that are not unique after the mapping from nucleotide to aminoacid, but that’s fine. Moreover if you pool data from multiple people you can still keep rearrangements that are found in multiple individuals because you are sure that they correspond to independent recombination events.
Note
Note about CDR3 sequence definition
This code is quite flexible, however it does demand a very consistent definition of CDR3 sequences.
CHECK THE DEFINITION OF THE CDR3 REGION OF THE SEQUENCES YOU INPUT. This will likely be the most often problem that occurs. The default models/genomic data are set up to define the CDR3 region from the conserved cysteine C (INCLUSIVE) in the V region to the conserved F or W (INCLUSIVE) in the J. This corresponds to positions X and X according to IMGT. This can be changed by altering the anchor position files, however the user is strongly recommended against this.